Ce test de dépistage des trisomies basé sur la présence d’ADN circulant, dérivant du placenta dans le sang maternel, a une excellente sensibilité. Il doit être associé à une consultation de conseil génétique pré-test pour permettre une décision éclairée.
Jusqu’à récemment, le dépistage prénatal non invasif des anomalies chromosomiques reposait sur le dosage d’HCG et PAPPA dans le sérum maternel, combiné à la mesure de la clarté nucale. Le taux de faux positifs engendré était d’environ 5 % et le taux de détection compris entre 80 et 95 % en fonction de la stratégie de dépistage utilisée. Les progrès technologiques de l’étude du génome ont permis la mise au point des techniques de dépistage prénatal non invasif (DPNI) basé sur la présence d’ADN circulant dérivé du placenta dans la circulation maternelle.
Le séquençage multiple en parallèle des fragments d’ADN maternel et placentaires (appelé aussi d’origine fœtale et intitulé fraction fœtale (FF) dans le sang maternel) est réalisé de manière conjointe. Le séquençage avec quantification peut être aléatoire ou focalisé et suivi par la quantification ou l’exploitation des polymorphismes d’un seul nucléotide. Les technologies de microarray (puce ADN ) peuvent aussi être utilisées pour quantifier l’ADN. L’analyse bioinformatique de ces méthodologies est complexe. Depuis l’introduction du DPNI en 2011, la place du DPNI dans le dépistage prénatal a fait l’objet de nombreuses évolutions. Toutes les techniques de dépistage génétique ont un risque résiduel de faux négatif. Ce sont donc bien des techniques de dépistage et non de diagnostic comme seul l’établissement du caryotype fœtal après amniocentèse ou biopsie de trophoblaste peuvent le permettre. Il s’agit cependant d’un dépistage à haute sensibilité puisque les taux de détection, en particulier en ce qui concerne la trisomie 21, sont supérieurs à 99 %.
Les recommandations de l’ACMG, American College of Medical Genetics and Genomics (Collège Américain de Génétique Médicale et de Génomique) (1), ont été publiées le 28 juillet 2016. Elles font suite aux premières recommandations édictées en 2013 et vont clairement dans le sens d’une extension des indications du dépistage non invasif, mais aussi du champ des syndromes dépistés.
Elles insistent sur deux points fondamentaux :
- les tests de dépistage sont des tests de dépistage et leur positivité doit être confirmée impérativement par un test invasif (amniocentèse ou biopsie de trophoblaste).
- la qualité de l’information délivrée aux patientes est fondamentale : elles doivent recevoir une information précise, adaptée, actualisée sur les différents types de dépistage disponibles et tout doit être fait de la part des prescripteurs comme des laboratoires pour développer des outils éducationnels sur le DPNI.
Les différentes études de validation aboutissent toutes aux mêmes conclusions : les taux de détection et la spécificité sont très élevés, autour de 99 % pour la trisomie 21, autour de 90 à 99 % pour la trisomie 18 et pour la trisomie 13 (2).
Importance d’une consultation de conseil génétique pré test
L’ACMG recommande, avant tout test non invasif, une consultation de conseil génétique expliquant le risque d’aneuploïdie, de translocation, microdélétion, et détaillant ce qui peut être dépisté ou non (anomalies monogéniques). Le but de ce conseil génétique est de faciliter la prise de décision par la patiente dès lors que lui ont été expliqués les résultats des examens déjà réalisés, les performances des tests disponibles en prenant leurs VPP et VPN (valeurs prédictives positives et négatives). La mise à disposition de ces informations permet aux patientes, au travers de leur propre système de valeurs, leurs convictions philosophiques, culturelles, ou religieuses, d’aboutir à une décision éclairée, dont les conséquences, limites, incertitudes doivent être clairement exposées. Après dépistage non invasif du premier trimestre, en l’absence de signe échographique anomal, les patientes doivent comprendre les avantages et les inconvénients de la détermination du caryotype fœtal et lui préférer la réalisation d’un prélèvement non invasif dont l’innocuité, le taux de détection élevé de trisomie 21, 13, 18, peuvent leur paraitre suffisants pour les rassurer, choisissant clairement de renoncer aux autres informations potentielles apportées par la détermination d’un caryotype complet.
Le délai de rendu des résultats a aussi une influence sur la décision de s’orienter vers une attitude invasive ou non invasive. En effet, l’étude de la clarté nucale et des marqueurs sériques dès 11/11 SA ½ permet en général un diagnostic par biopsie de villosité choriale à partir de 12/12 SA ½ et quand une anomalie est diagnostiquée, les parents qui désirent avoir recours à une IMG peuvent encore y avoir accès par aspiration. Il faut donc être vigilant à ce que l’évolution prévisible vers un diagnostic non invasif proposé à toutes les femmes fasse appel à des tests de délai de réponse rapide (8/10 jours maximum) et soit accessible tôt dans la grossesse afin de ne pas pénaliser les patientes par une prise éventuelle de décision d’IMG après 15/16 SA. En cas d’anomalies morphologiques ou d’hyperclarté nucale supérieure à 3.5 voire 3 mm, les patientes doivent comprendre que si l’on désire avoir un diagnostic chromosomique le plus précis possible, la réalisation d’un caryotype avec une analyse chromosomique en CGH Array (ACPA) est nécessaire. Cette solution va permettre de diagnostiquer au prix d’un risque de complications très faible (0,1-0,2 % dans des mains entrainées) (3) toutes les aneuploidies, les anomalies chromosomiques déséquilibrées, les gains ou pertes de matériel chromosomiques de petite taille (Copy Number variants ou CNV).
Le dépistage non invasif doit-il être proposé chez les patientes à faible risque ou à risque intermédiaire ?
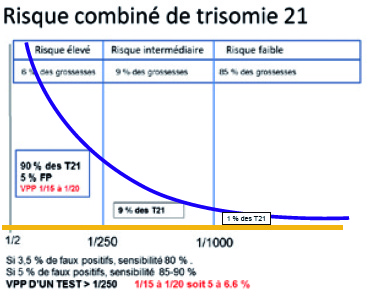 Le schéma ci-contre illustre la situation obtenue après évaluation du risque combiné de trisomie 21. 5 à 6 % des patientes sont à risque supérieur à 1/250 et, dans cette catégorie, sont regroupés environ 90 % des cas de trisomie 21. Les patientes présentant un risque combiné de trisomie 21 compris pour le risque intermédiaire entre 1/250 et 1/1000 (environ 9 à 10 % des patientes et 9 % des trisomies 21), et pour le risque faible en deçà de 1/1000 (correspondant à 85 % des grossesses et environ 1 % des fœtus porteurs de trisomie 21). L’ACMG recommande d’informer toutes les femmes enceintes du fait que le DPNI représente l’option de dépistage des trisomies 21, 13,18 dotée de la plus grande sensibi-lité. Dans les situations de risque intermédiaire, la pratique d’un dépistage non invasif initialement décriée sans fondement rationnel permet bien évidemment d’augmenter le taux de dépistage des principales aneuploïdies avec un taux de détection et un taux de faux positifs inchangés. Il faut cependant noter qu’à partir du moment où la prévalence d’une affection diminue dans la population étudiée, la valeur prédictive d’un test positif diminue. Alors qu’en population à haut risque (supérieur à 1/250), la valeur prédic-tive d’un test non invasif est supérieure à 90 voire 99 %, en population générale, elle ne dépasse pas 50 %. Elle demeure cependant bien supérieure à la VPP d’un test combiné du premier trimestre de la grossesse, qui ne dépasse pas 5 à 6 %.
Le schéma ci-contre illustre la situation obtenue après évaluation du risque combiné de trisomie 21. 5 à 6 % des patientes sont à risque supérieur à 1/250 et, dans cette catégorie, sont regroupés environ 90 % des cas de trisomie 21. Les patientes présentant un risque combiné de trisomie 21 compris pour le risque intermédiaire entre 1/250 et 1/1000 (environ 9 à 10 % des patientes et 9 % des trisomies 21), et pour le risque faible en deçà de 1/1000 (correspondant à 85 % des grossesses et environ 1 % des fœtus porteurs de trisomie 21). L’ACMG recommande d’informer toutes les femmes enceintes du fait que le DPNI représente l’option de dépistage des trisomies 21, 13,18 dotée de la plus grande sensibi-lité. Dans les situations de risque intermédiaire, la pratique d’un dépistage non invasif initialement décriée sans fondement rationnel permet bien évidemment d’augmenter le taux de dépistage des principales aneuploïdies avec un taux de détection et un taux de faux positifs inchangés. Il faut cependant noter qu’à partir du moment où la prévalence d’une affection diminue dans la population étudiée, la valeur prédictive d’un test positif diminue. Alors qu’en population à haut risque (supérieur à 1/250), la valeur prédic-tive d’un test non invasif est supérieure à 90 voire 99 %, en population générale, elle ne dépasse pas 50 %. Elle demeure cependant bien supérieure à la VPP d’un test combiné du premier trimestre de la grossesse, qui ne dépasse pas 5 à 6 %.
Doit-on chercher autre chose que les trisomies 13, 18 et 21 ?
Depuis plusieurs années, les laboratoires spécialisés dans le dépistage non invasif ont mis au point des outils permettant de dépister d’autres pathologies que les trisomies 21, 13, et 18. Ceci peut inclure le dépistage des trisomies 16 et 22, des anomalies des chromosomes sexuels, de nombreuses microdélétions impliquées dans des pathologies foetales graves, des délétions supérieures à 7 Mb sur chaque chromosome. Ces dépistages sont donc désormais disponibles et validés : Lefkowitz et coll, Am J Obstet Gynecol. 2016 (4) ont étudié de manière rétrospective 1222 prélèvements en aveugle, issus de patientes à haut risque d’anomalies chromosomiques finalement classifiées en trisomies 21, 13, 18, anomalies des chromosomes sexuels, duplications ou délétions > 7mb, et plusieurs délétions > 7 Mb. Les sensibilités retrouvées étaient de 100 % pour les T21 (intervalle de confiance à 95 % [95 % CI], 94.6-100 %), T18 (95 % CI,84.4-100 %), T13 (95 % CI, 74.7-100 %), et anomalies des chromosomes sexuels (95 % CI, 84-100 %) et de 97.7 % pour les anomalies > 7 Mb (95 % CI, 86.2-99.9 %), avec des spécificités de 100 % pour les T21 (95 % CI,99.6-100 %), T18 (95 %CI, 99.6100 %), et T13 (95 %CI, 99.6-100 %), et 99.9 % les anomalies des chromosomes sexuels et les anomalies > 7 Mb (95 % CI, 99.4-100 %).
Doit-on dépister les anomalies des chromosomes sexuels ?
Il est important de noter que l’ACMG conseille d’informer toutes les femmes enceintes sur la possibilité de réaliser un dépistage des anomalies des chromosomes sexuels par étude de l’ADN fœtal dans le sang maternel. S’il existe un consensus unanime sur le fait d’éviter que ce type de dépistage ne puisse servir à une demande d’identification du sexe fœtal à visée de sélection du sexe qui est désiré par les parents, il n’y a pas de consensus sur la nécessité de dépister les syndromes de Turner et de Klinefelter et les trisomies X.
De nombreuses interrogations éthiques concernent la possibilité de dépister un syndrome de Turner ou un syndrome de Klinefelter en anténatal sur l’ADN fœtal dans la circulation
maternelle. Il est légitime de se demander en quoi il serait moins éthique de les dépister sur un test non invasif et de confirmer le diagnostic par amniocentèse, alors que ces anomalies sont
mises en évidence quotidiennement lors de l’établissement du caryotype foetal, et que de nombreuses informations peuvent être apportées aux couples confrontés à ce diagnostic.
Il existe de nombreux risques potentiels dans le cas du dépistage des anomalies des chromosomes sexuels, à commencer par l’anxiété parentale, le risque de discrimination ou de regard différent des parents, amis, société, la possibilité de demandes d’IMG. Quand le diagnostic est fortuit, une demande d’IMG est exprimée dans environ 11 à 15 % des cas, le recours systématique à un généticien ou à un pédiatre spécialisé permettant d’en diminuer les demandes.
A contrario, faire appel à des groupes de discussion, parler à l’enfant de sa particularité chromosomique peut être un processus graduel sur plusieurs années avec aide de professionnels
et est susceptible de lui donner accès à une prise en charge plus adaptée. Un diagnostic précoce permet la mise en place de stratégies compensatrices visant à améliorer le pronostic : orthophonie vers l’âge de 5 ans, psychomotricité, prise en charge par psychologues, pédiatres, endocrinologues (consultation tous les 3 mois / Hôpital de jour tous les ans) 80 % des patients présentant un syndrome de Turner ne présentent pas de déficit intellectuel, les autres ayant un retard de 5 à 10 points de QI par rapport à la fratrie, avec un QI verbal nettement supérieur au QI performances (repérage, arithmétique…).
Dans 20 % des cas, une scolarité adaptée est nécessaire et il existe un manque d’autonomie, des difficultés d’orientation temporo-spatiale, de mémorisation, d’attention, de coordination motrice, en mathématiques, un retrait dans la vie sociale…
Si retard mental est associé, il est nécessaire de chercher un autre remaniement chromosomique associé. Les patients atteints de syndrome de Klinefelter présentent un déficit intellectuel modéré avec un QI moyen compris entre 85 et 90 (-20 par rapport à la fratrie), un QI performance supérieur au QI verbal, des difficultés à écouter, à enregistrer les consignes, un déficit de mémoire auditive. Un retard d’acquisition de la lecture, une labilité de l’attention, immaturité, sentiment d’insécurité, timidité, défaut de jugement, difficultés à se référer aux pairs, constituent une fois sur 2 le motif de consultation. L’ajustement psycho-social difficile est rencontré dans : 30-40 % des cas. Les bénéfices potentiels du screening des anomalies des chromosomes sexuels sont nombreux ; un diagnostic précoce peut permettre une prise en charge plus précoce et une meilleure prise en charge. Dans le cas d’un syndrome de Turner, l’administration de GH peut permettre d’’augmenter la taille finale ; l’identification d’une cardiopathie permet une prise en charge anténatale plus adaptée.
Dans le cas d’un syndrome de Klinefelter, il est important de diminuer le risque d’ostéoporose et de tenter d’augmenter la fertilité. Un traitement substitutif par la testostérone dès
11-12 ans dès que le dosage de testostérone est inférieur à la normale et que le taux de gonadotrophines est élevé permet d’obtenir une apparence physique plus masculine, une augmentation de la pilosité (visage et pubis), une amélioration de l’estime de soi, une diminution de la fatigue et de l’irritabilité, une augmentation de la libido, de la force et de la densité osseuse. Une consultation dans un CECOS vers 15-16 ans peut être discutée pour tenter une cryoconservation de spermatozoïdes.
Doit-on dépister les microdélétions et les CNV ?
Les microdélétions peuvent induire des anomalies physiques et neuro-développementales au moins aussi sévères que les aneuploïdies; leur risque est indépendant de l’âge maternel. On considère qu’il existe des microdélétions et des micro-duplications à traduction clinique dans 1 à 1.7 % des grossesses, leur risque étant similaire à celui de trisomie 21 chez les patientes de moins de 30 ans alors que leur pronostic intellectuel est moins bon que celui de la T21. Leur diagnostic échographique est par ailleurs difficile, peu spécifique.
Leur revue en détail en justifie le dépistage :
– Le syndrome de Di George causé par la délétion 22q11.2 est une anomalie chromosomique congénitale fréquente, dont la prévalence est de 1/2 000. Une délétion de 3 Mb contribue pour 87 % de l’ensemble des DGS (Di George Syndrome). Cette anomalie est caractérisée le plus souvent par des malformations cardiaques et palatines, une dysmorphie faciale, un retard du développement et une immunodéficience. Le syndrome de délétion 22q11.2 présente un phénotype clinique qui varie de modéré à sévère.
Le DGS est commun et représente la 2e cause de malformation cardiaque congénitale, plus commun dans la tétralogie de Fallot que la T21, la 1ère cause d’anomalies du palais (fentes palatines, labiales, palatines sous-muqueuse, dysfonction vélo-pharyngée). Il représente également la 2e cause de retard mental après la trisomie 21 et est impliqué dans environ 2.4 % des retards mentaux.
Cause fréquente de troubles d’apprentissage, le syndrome de Di George a une morbidité significative : touche tous les organes (problèmes immunitaires, endocrines, gastro-intestinaux) – déficits cognitifs variables et troubles psychiatriques. Son risque n’est pas corrélé à l’âge maternel. Sa grande variabilité peut empêcher un diagnostic précoce et est susceptible d’aggraver le pronostic. Son diagnostic anténatal est difficile puisque 30 % des fœtus n’ont pas de cardiopathie ni de fente palatine et 50 % n’ont pas de malformation rénale.
Poser le diagnostic d’un syndrome de Di George va permettre préparation et programmation aussi bien sur les plans médical que psychologique et également une réduction des coûts liés aux diagnostics tardifs ou non faits. Or, 70 à 90 % ont des difficultés d’apprentissage et 25 % seront schizophrènes à l’âge adulte.
Nécessité d’une prise en charge multidisciplinaire toute la vie, à l’origine de difficultés de vies au quotidien, justifiant une prise en charge précoce et adaptée, seule susceptible de réduire la morbidité du syndrome. Les performances du dépistage non invasif ont été modélisées par Wapner et coll (5) avec une valeur prédictive positive de 5.3 % et une valeur prédictive négative > 99.99 %, et sont en cours d’étude clinique.
– Les délétions 15q11q12 sont à l’origine des syndromes de Prader-Willi et d’Angelman dont la prévalence est de 1/20000. 70 % des syndromes de Prader-Willi sont secondaires à des délétions 15q paternelles; 60 à 75 % syndromes d’Angelman s’expliquent par des délétions 15q maternelles. Le retard mental est constant pour ces deux affections. Le diagnostic échographique du syndrome d’Angelman est impossible. Pour le Prader-Willi, l’hypomobilité est un signe d’appel mais peu spécifique donc il n’y quasiment pas de diagnostic décrit en anténatal.
– Les délétions 1p, 4p, 5p s’accompagnent souvent d’une hypotrophie ; la dysmorphie faciale est peu accessible aux échographies de routine et le pronostic intellectuel est catastrophique pour les 3 anomalies, beaucoup plus sévère que dans le cas de la T21. Ces anomalies peuvent être isolées mais également dans le cadre de translocation avec risque de récidive (translocation parentale équilibrée se déséquilibrant à la génération suivante).
– Les trisomie 16 et trisomie 22 représentent la première cause de trisomie à la conception.
Elles sont pourvoyeuses d’avortements spontanés, le plus souvent précoces, mais pouvant se produire jusqu’à 15-16 SA, d’où l’intérêt de les dépister à 12 SA. Des trisomies 22 homogènes sont rapportées chez quelques enfants vivants avec difficultés d’apprentissages. Des trisomies 16 confinées au placenta, sous formes de mosaïques, se compliquent classiquement de retards de croissance intra-utérins sévères. La notion d’un résultat positif de dépistage non invasif de trisomie 16 doit entrainer la réalisation d’une amniocentèse complémentaire et de mettre en place une surveillance échographique spécifique. Identifier l’étiologie de tels retards de croissance permettra bien évidement la réalisation d’un conseil génétique adapté.
Le dépistage prénatal des aneuploïdies évolue rapidement vers un dépistage complet, basé sur échographie, marqueurs sériques, DPNI, étude du caryotype, analyse chromosomique par puce ADN.
L’échographie garde toute sa place pour valider les résultats d’analyse sur trophoblaste direct, s’assurer de l’absence d’anomalie anatomique dans le cadre d’un DPNI, aider à l’évaluation pronostique et thérapeutique des anomalies anatomiques, ou des syndromes malformatifs. Sans conteste dans la zone ½ à 1:250 le DPNI permet d’éviter 96 % des prélèvements et, dans les risques intermédiaires, il est désormais justifié d’informer les patientes de la possibilité de DPNI et de l’intérêt des échographies orientées. En concordance avec les recommandations de l’ACMG, toutes les patientes pourraient être informées des possibilités de dépistage génétique étendu aux microdélétions, anomalies des chromosomes sexuels, trisomies 16 et 22, anomalies supérieures à 7 Mb en étude pan-génomique. Dans certaines situations obstétricales difficiles et échographiques complexes, le DPNI permet d’éviter un prélèvement invasif potentiellement dangereux tout en diminuant considérablement le risque résiduel d’aneuploïdie. Ces situations doivent bien sur être discutées au sein des structures spécialisées des centres pluridisciplinaires de diagnostic prénatal.
L’information pré-test est fondamentale et ne saurait se résumer à un formulaire de consentement signé à la va-vite. Elle doit exposer le spectre clinique des anomalies recherchées,
toujours donner la possibilité de ne pas opter pour un dépistage étendu, et a pour but d’aider les patientes à choisir le dépistage qui correspond le mieux à leurs attentes, leur
permettant un consentement éclairé autonome.
Le développement des techniques de dépistage va enfin conduire à s’interroger sur les dépistages de porteurs sains en population (carrier screening) et les implications éthiques de ces nouveaux développements.
François JACQUEMART, Géraldine VIOT, Philippe BOUHANNA – Centre pluridisciplinaire de Diagnostic Prénatal, Hopital Américain de Paris, Neuilly sur Seine
Les auteurs déclarent n’avoir aucun lien d’intérêt pour cet article.
RÉFÉRENCES
- Gregg AR, Skotko BG, Benkendorf JL, Monaghan KG, Bajaj K, Best RG, Klugman S, Watson MS. Noninvasive prenatal screening for fetal aneuploidy, 2016 update: a position statement of the American College of Medical Genetics and Genomics. Genet Med. 2016 Jul 28. doi: 10.1038/gim.2016.97. [Epub ahead of print]
- Gil MM, Quezada MS, Revello R, Akolekar R, Nicolaides KH. Analysis of cell-free DNA in maternal blood in screen ing for fetal aneuploidies: updated meta-analysis. Ultrasound Obstet Gynecol. 2015 Mar;45(3):249-66. doi: 10.1002/uog.14791. Epub 2015 Feb 1.
- Akolekar R, Beta J, Picciarelli G, Ogilvie C, D’Antonio F. Procedure-related risk of miscarriage following amniocentesis and chorionic villus sampling: a systematic review and meta-analysis.Ultrasound Obstet Gynecol. 2015 Jan;45(1):16-26. doi: 10.1002/uog.14636.
- Lefkowitz RB, Tynan JA, Liu T, Wu Y, Mazloom AR, Almasri E, Hogg G, Angkachatchai V, Zhao C, Grosu DS, McLennan G, Ehrich M. Clinical validation of a noninvasive prenatal test for genomewide detection of fetal copy number variants. Am J Obstet Gynecol. 2016 Aug;215(2):227.e1-227.e16. doi:10.1016/j.ajog.2016.02.030. Epub 2016 Feb 17
- Wapner RJ, Babiarz JE, Levy B, Stosic M, Zimmermann B, Sigurjonsson S, Wayham N, Ryan A, Banjevic M, Lacroute P, Hu J, Hall MP, Demko Z, Siddiqui A, Rabinowitz M, Gross SJ, Hill M, Benn P. Expanding the scope of noninvasive prenatal testing: detection of fetal microdeletion syndromes. Am J Obstet Gynecol. 2015 Mar;212(3):332.e1-9. doi: 10.1016/j.ajog.2014.11.041. Epub 2014 Dec 2.





42 commentaires